Cri du Chat Sendromu

Cri du Chat Sendromu
Whatsapp Facebook Twitter LinkedIn
{kind=link}
Cri du chat sendromu ya da diğer adıyla kedi miyavlaması sendromu; kromozom 5’in kısa kolunun değişken bir kısmının eksik veya silinmiş monozom) olduğu nadir bir genetik hastalıktır. Belirtiler, silinen genetik maddenin tam boyutuna ve konumuna bağlı olarak durumdan duruma değişiklik gösterir. Yaygın semptomlar arasında bir kedinin sesine benzeyen ayırt edici bir çığlık, karakteristik yüz özellikleri, yavaş büyüme ve mikrosefali vardır. Bu urum, baş çevresinin bir çocuğun yaşı ve cinsiyeti için beklenenden daha küçük olduğunu gösteren bir durumdur. Etkilenen çocuklar, kas ve zihinsel faaliyetlerin koordinasyonunu gerektiren ve orta ila şiddetli zihinsel özürlülük gerektiren becerilerin kazanılmasında gecikmeler de gösterir. Vücudun farklı organ sistemlerini etkileyen ek semptomlar da ortaya çıkabilir. Bu bozukluk ilk olarak 1963 yılında tıp literatüründe, ayırt edici kedi benzeri ağlamadan sonra hastalığa isim veren Doktor Lejeune tarafından tanımlanmıştır. Fransızca’da, Cri du sohbeti kedinin ağlaması şeklinde çeviri yapılmaktadır.
Belirtiler
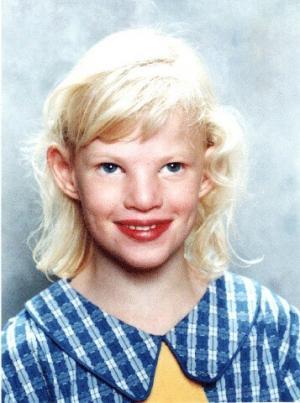
Cri du chat sendromunun semptomları vakadan duruma değişir. Cri du chat sendromu ile ilişkili karakteristik olarak yaşamın ilk birkaç haftasında yüksek perdeli, keskin ağlama mevcuttur. Bir kedinin pervazına benzeyen ağlama, etkilenen bebekler büyüdükçe daha az belirginleşir. Etkilenen bebekler ayrıca düşük doğum ağırlığı, büyüme eksiklikleri, azalan kas tonusu ve mikrosefali gösterebilir; bu durum, baş çevresinin bir çocuğun yaşı ve cinsiyeti için beklenenden daha küçük olduğunu gösteren bir durumdur.[ cri du 1]
Belirgin yüz özellikleri arasında anormal şekilde yuvarlak veya dolgun bir yüz, geniş bir burun köprüsü, geniş aralıklı gözler, çapraz gözler şaşılık), aşağı doğru eğimli göz kapakları kıvrımları, göz kapaklarını tutabilen dikey cilt kıvrımları olabilir. İç köşeler, düşük ayarlanmış kulaklar ve anormal derecede küçük bir çene mevcuttur, üst ve alt dişlerin yanlış hizalanması da oluşabilir.
Ek yüz özellikleri arasında, üst dudaktan buruna burnun anormal derecede küçük bir mesafesi, ağzın çatısının tam kapanmaması yarık damak), üst dudakta anormal bir yiv veya boşluk yarık dudak) ve anormal dolgunluk sayılabilir. Alt dudağın ek olarak, boğazın arkasına sarkan etli kütle dökülebilir. Etkilenen bebekler yaşlandıkça yüz dolgunluğunu kaybedebilir ve anormal derecede uzun ve dar hale gelebilir.
Etkilenen bebeklerin çoğu ayrıca bir dereceye kadar psikomotor ve zihinsel sakatlık sergilemektedir. Psikomotor sakatlık, kafa kontrolü, oturma ve yürüme gibi zihinsel ve kaslı aktiviteler gerektiren becerilerin kazanılmasında gecikmedir. Cri du chat sendromu olan çocukların yaklaşık yarısı 5 yaşına kadar kendileri giyinebilirler. Çoğu durumda orta ila şiddetli zihinsel yetersizlik mevcuttur. Konuşma gelişimi özellikle cri du chat sendromu olan çocuklarda gecikir. Etkilenen çocuklar konuşmayı genellikle iletişim kurabildiklerinden daha iyi anlarlar. Bazı çocuklar hiperaktivite veya kendi kendine kötü niyetli davranışlar gösterebilir. Cri du chat sendromlu çocuklar hipotonik düşük kas tonu) doğar, yaşlandıkça hipertonik yüksek kas tonu) olma eğilimindedirler.
Etkilenen bebekler düşük kas tonusu, emme sorunları ve gastroözofageal reflü hastalığı nedeniyle beslenme güçlüğü çekebilir. Bazıları ayrıca zatürrelere yol açabilecek aspirasyon riski altındadır. Bir çalışmada, cri du chat sendromlu çocukların sadece% 50’si 3,5 yaşına kadar bir kaşıkla kendileri beslenebilmiş oldukları tespit edilmiştir. Cri du chat sendromu ile ilişkili olarak çeşitli ek bulgular ortaya çıkabilir. Omurganın anormal yan yana eğriliği skolyoz) sık görülen bir komplikasyondur. Etkilenen çocuklar aynı zamanda kulak enfeksiyonu ve işitme kaybı riski taşırlar. Etkilenen bebeklerin yaklaşık yüzde 15-20’sinde doğuştan kalp defekti vardır. En sık görülen kalp defekti, akciğerlere ve vücudun ana atardamarı aort), doğumdan sonra kapanamamasına neden olan bir durum olan duktus arteriosus’tur.
Cri du chat sendromu ile ilgili daha az rastlanan bulgular, alt karın kasık fıtığı) destek dokusunda bağırsakların bir kısmının dışarı çıkmasına izin veren bir yırtığın gelişmesi, mide veya ince bağırsakların içeriğinin yemek borusuna geçişi veya geri akması, böbrek ve idrar yolu anormallikleri, solunum güçlüğüdür. El ve ayak parmaklarında bir parmağın dördüncü parmağa doğru içe doğru anormal şekilde bükülmesi veya eğrilmesi görülebilir. Ayrıca PEV ve ses kutusunun yapısal anomalileri gırtlak) görülebilir. Bazı durumlarda yakın görüşlülük ve katarakt gelişebilir. Ayrıca saçın erken beyazlaması da rapor edilmiştir. Bazı kişiler tekrarlayan solunum ve bağırsak enfeksiyonları geliştirebilir. Etkilenen erkek bebeklerde, testisler skrotuma inemez ve idrar açıklığı penisin altına yerleştirilebilir. Ayrıca, cri du chat ve Hirschsprung hastalığı ile bir ilişkisi olduğu tespit edilmiştir.
Nedenleri
 Cri du chat sendromu, kromozom 5’in kısa kolunun p) değişen uzunluğu olan kısmi bir silme nedeniyle meydana gelen kromozomal bir hastalıktır. İnsan kromozomlarının çiftleri 1 ila 22 arasında numaralandırılmıştır ve erkeklerde bir X ve bir Y kromozomu ve kadınlarda iki X kromozomu içeren ek bir 23. cinsiyet kromozomu çiftidir. Her kromozomun p işaretli kısa bir kolu ve q işaretli uzun bir kolu vardır.
Cri du chat sendromu, kromozom 5’in kısa kolunun p) değişen uzunluğu olan kısmi bir silme nedeniyle meydana gelen kromozomal bir hastalıktır. İnsan kromozomlarının çiftleri 1 ila 22 arasında numaralandırılmıştır ve erkeklerde bir X ve bir Y kromozomu ve kadınlarda iki X kromozomu içeren ek bir 23. cinsiyet kromozomu çiftidir. Her kromozomun p işaretli kısa bir kolu ve q işaretli uzun bir kolu vardır.
Kromozomlar ayrıca numaralandırılmış birçok gruba bölünmüştür. Örneğin, kromozom 5p15.3, kromozom 5’in kısa kolundaki bant 15 ile ilgilidir. Numaralı bantlar, her bir kromozomda bulunan binlerce genin konumunu belirtir. Cri du chat sendromu olan bireylerde, ilişkili semptomların ve bulguların kapsamı ve ciddiyeti, kromozom 5p’nin silinmiş kısmının tam uzunluğuna veya konumuna bağlı olarak değişebilir. Araştırmacılar, belirli semptomların kromozom 5’in kısa kolundaki spesifik bölgelerle ilişkili olabileceğini belirlemişlerdir. Araştırmacılar, cri du chat sendromunun gelişiminde rol oynadığına inanılan birkaç gen tanımladılar. Telomeraz ters transkriptazbant 13.33 5p13.33) kromozom 5 kısa kolu üzerinde yer almaktadır geni ve5p15.2’deki semafor F geni, çeşitli özelliklere katkıda bulunabilir. D-katenin geninin, ayrıca 5p15.2’de silinmesi, bu protein erken nöronal gelişimde ifade edildiğinden, daha ciddi zihinsel engellilik ile bağlantılıdır. Araştırmacılar belirli belirti ve bulgular kümelerini kromozom 5p’nin silinmesine bağlayabilirse, tanı ve prognoza büyük ölçüde yardımcı olabilir.
Çoğu cri du chat sendromu vakası, embriyonik gelişimde çok erken bilinmeyen nedenlerle kendiliğinden ortaya çıkmaktadır. Çoğu silmeler % 80-90) menşeilidir, yani sperm oluşumunun bir parçası olarak ortaya çıkarlar. De novo delesyonu olan bir çocuğun ebeveynleri genellikle normal kromozomlara sahiptir ve kromozomal anomalisi olan başka bir çocuğa sahip olma riski nispeten düşüktür.
Olguların yaklaşık yüzde 10-15’inde, cri du chat sendromu, 5p kromozomu ve başka bir kromozom veya kromozom içeren dengeli bir translokasyondan kaynaklanabilir. Translokasyonlar, bazı kromozom bölgeleri kopup yeniden düzenlendiğinde meydana gelir, bu da genetik materyalin ve değiştirilmiş bir kromozom setinin kaymasına neden olur. Bu tür translokasyonlar, bilinmeyen nedenlerle kendiliğinden meydana gelebilir veya böyle bir dengeli translokasyonun taşıyıcısı olan bir ebeveyn tarafından iletilebilir. Dengeli bir yer değiştirme, değiştirilmiş fakat dengelenmiş bir kromozom grubundan oluşur ve genellikle taşıyıcıya zararsızdır. Bununla birlikte, böyle bir kromozomal yeniden düzenleme, taşıyıcının yavrularında artan bir anormal kromozomal gelişme riski ile ilişkili olabilir. Kromozomal analiz, ebeveynin dengeli bir translokasyonu olup olmadığını belirleyebilir.
Etkilenen Kişiler
Cri du chat sendromu kadınları erkeklerden daha sık etkiler. İnsidansı 1-15.000 ila 50.000 canlı doğum arasında değişmektedir. Bazı cri du chat sendromu vakaları genel popülasyonda bu hastalığın gerçek sıklığını belirlemeyi zorlaştırarak teşhis edilmeyebilir.
İlgili Bozukluklar
 Aşağıdaki bozuklukların belirtileri cri du chat sendromu ile benzer olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir. Wolf sendromu olarak da bilinen Wolf-Hirschhorn sendromu, kromozom 4’ün 4p) kısa kolunun p) kısmi olarak silinmesi olduğu nadir bir kromozomal hastalıktır. 4p silinmesinin büyüklüğü ve konumu durumdan duruma değişse de, 4p16.3 bandının silinmesinin, bozukluğun karakteristik özelliklerine yol açan kritik bölge olduğuna inanılmaktadır. İlişkili anormallikler tipik olarak düşük doğum ağırlığı, büyüme geriliği, zayıf kas tonusu ve fiziksel ve zihinsel aktivitelerin koordinasyonunu gerektiren becerilerin kazanılmasında gecikmeleri içerir. Etkilenen bebeklerin ve çocukların çoğu, kafatası ve yüz bölgesinde belirgin malformasyonlara sahiptir. Bunlar arasında küçük bir kafa ve yüksek alın, yüksek kemerli kaşlar, geniş aralıklı gözler, gözlerin iç köşelerini örten dikey cilt kıvrımları, anormal derecede geniş bir burun köprüsüne sahip gagalı bir burun, kapalı bir ağız, üst dudağın ortasındaki alışılmadık şekilde kısa dikey bir oluk veya büyük, hatalı biçimlendirilmiş kulaklar görülebilir. Bunlar veya ilave kraniyofasiyal malformasyonlar nedeniyle, yüz bir taraftan diğerine nispeten farklı görünebilir. Ek fiziksel anormallikler de mevcut olabilir.
Aşağıdaki bozuklukların belirtileri cri du chat sendromu ile benzer olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir. Wolf sendromu olarak da bilinen Wolf-Hirschhorn sendromu, kromozom 4’ün 4p) kısa kolunun p) kısmi olarak silinmesi olduğu nadir bir kromozomal hastalıktır. 4p silinmesinin büyüklüğü ve konumu durumdan duruma değişse de, 4p16.3 bandının silinmesinin, bozukluğun karakteristik özelliklerine yol açan kritik bölge olduğuna inanılmaktadır. İlişkili anormallikler tipik olarak düşük doğum ağırlığı, büyüme geriliği, zayıf kas tonusu ve fiziksel ve zihinsel aktivitelerin koordinasyonunu gerektiren becerilerin kazanılmasında gecikmeleri içerir. Etkilenen bebeklerin ve çocukların çoğu, kafatası ve yüz bölgesinde belirgin malformasyonlara sahiptir. Bunlar arasında küçük bir kafa ve yüksek alın, yüksek kemerli kaşlar, geniş aralıklı gözler, gözlerin iç köşelerini örten dikey cilt kıvrımları, anormal derecede geniş bir burun köprüsüne sahip gagalı bir burun, kapalı bir ağız, üst dudağın ortasındaki alışılmadık şekilde kısa dikey bir oluk veya büyük, hatalı biçimlendirilmiş kulaklar görülebilir. Bunlar veya ilave kraniyofasiyal malformasyonlar nedeniyle, yüz bir taraftan diğerine nispeten farklı görünebilir. Ek fiziksel anormallikler de mevcut olabilir.
Bu özellikler arasında bir gözün diğerine göre şaşılık) anormal sapması olabilir, gözün renkli bölgesinden kısmi doku yokluğu, ağız çatısının tam kapanmaması yarık damak), inmemiş testisler ve etkilenen erkeklerde penisin altına idrar açıklığının anormal yerleştirilmesi, kalbin yapısal bozuklukları, beyindeki kontrolsüz elektriksel aktivitenin ani bölümleri nöbetler), iskelet anomalileri veya diğer bulgulara da rastlanabilir. Wolf-Hirschhorn sendromu genellikle embriyonik gelişimde çok erken bilinmeyen nedenlerle kendiliğinden ortaya çıkmaktadır. Daha az yaygın olarak, ebeveynlerden birinde dengeli bir yer değiştirme sonucu ortaya çıkabilir.
Ek kromozomal bozukluklar, cri du chat sendromu ile ilişkili özelliklere benzer özelliklere sahip olabilir. Mevcut spesifik kromozomal anomaliyi doğrulamak için kromozomal test gereklidir. Bu tür bozukluklar hakkında daha fazla bilgi için, söz konusu spesifik kromozomal bozukluğun adını seçilerek veya nadir olan hastalık veritabanında arama terimi olarak kromozom kullanılmalıdır.
Teşhis
Yeni doğanlarda, cri du chat sendromu tanısı, kapsamlı bir klinik değerlendirme, karakteristik bulguların örn. Kedi benzeri ağlama) tanımlanması ve kromozom 5’in kısa kolunda delesyon ortaya çıkaran kromozomal çalışmalar ile doğrulanır. Floresan in situ hibridizasyon FISH) olarak bilinen test, cri du chat sendromu tanısını doğrulamak için kullanılabilir.
Bir ebeveynde dengeli bir translokasyonun olup olmadığını belirlemek için kromozomal çalışmalar da yapılabilir. Skolyoz gibi iskelet anormalliklerini ortaya çıkarmak için röntgen gibi rahatsızlıkların derecesini belirlemek için ek tanısal testler kullanılabilir. Kromozomal anormalliklerin belirlenmesinde bilimsel teknikler gittikçe daha fazla rafine edilmektedir. Bu, tanı tekniklerinin düzeldiği ve bazı durumlarda cri du chat sendromunun doğum öncesi tanısının mümkün olduğu anlamına gelir.
Tedavi
Cri du chat sendromunun tedavisi, her bir bireyde belirgin olan spesifik semptomlara 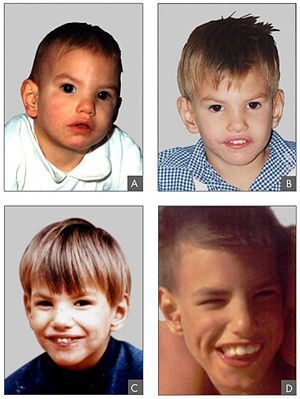 yöneliktir. Tedavi, uzman bir ekibin koordine çabalarını gerektirebilir. Çocuk doktorları, ortopedistler, cerrahlar, kardiyologlar, konuşma patologları, nörolog, diş hekimi, fiziksel ve mesleki terapistler,diğer sağlık uzmanlarının etkilenen bir çocuğun tedavisini sistematik ve kapsamlı bir şekilde planlaması gerekebilir. Cri du chat yapan bazı çocukların duyusal-sinirsel sağırlığı olabileceğinden, işitsel testler yapılmalıdır.
yöneliktir. Tedavi, uzman bir ekibin koordine çabalarını gerektirebilir. Çocuk doktorları, ortopedistler, cerrahlar, kardiyologlar, konuşma patologları, nörolog, diş hekimi, fiziksel ve mesleki terapistler,diğer sağlık uzmanlarının etkilenen bir çocuğun tedavisini sistematik ve kapsamlı bir şekilde planlaması gerekebilir. Cri du chat yapan bazı çocukların duyusal-sinirsel sağırlığı olabileceğinden, işitsel testler yapılmalıdır.
Erken dönem bu sendrom olan çocukların, en yüksek potansiyele ulaşmalarını sağlamada önemlidir. Yararlı olabilecek hizmetler arasında özel iyileştirici eğitim, fizik tedavi, konuşma terapisi, özel servisler ve diğer tıbbi, sosyal veya mesleki hizmetler yer alabilir. Çocukların çoğu, bir yaşından önce tedaviye kayıtlıdır.
Konjenital kalp defektleri, şaşılık, skolyoz, kuluçka ayakları, yarık damak ve dudak da dahil olmak üzere cri du chat sendromu ile ilişkili çeşitli semptomların tedavisi için cerrahi uygulanabilir. Cri du chat olan çocuklar için hayatta kalmak genellikle iyidir. Sendrom ile ilgili ölümlerin çoğu yaşamın ilk yılında meydana gelmekte ve birkaç çocuk 50 yaşın üzerinde yaşamıştır. Etkilenen bireyler ve aileleri için genetik danışmanlık tavsiye edilir. Diğer tedavi semptomatik ve destekleyicidir.
Araştırma Terapileri
Cri du chat sendromu araştırmaları ve çalışmaları devam etmektedir. Bir araştırma, erken yaşta özel okullaşma, ev ortamı kurumsal ortam yerine) ve aile desteğinin, hastanın normal bir beş veya altı yaşında birinin yeteneklerini kazanmasına yardımcı olabileceğini göstermiştir. Aynı çalışmada, özel eğitim görmüş ve destekleyici bir ev ortamında yaşayan on yaşın üzerindeki çocukların yarısı yeterince iletişim kurabilir durumda olduğu gözlemlenmiştir.
Kaynakça:
ghr.nlm.nih.gov
emedicine.medscape.com
https://rarediseases.org